
Research
Our £500 million investment into research since 1960 has helped transform treatments and taken us to the point where beating blood cancer is now in sight. Our researchers are working to finish the job.
Research blogs
Learn more about our blood cancer research and for regular updates on what we know about the effectiveness of covid vaccines for people with blood cancer.

The coronavirus vaccine: what you need to know

Why we fund Research
Our research impact
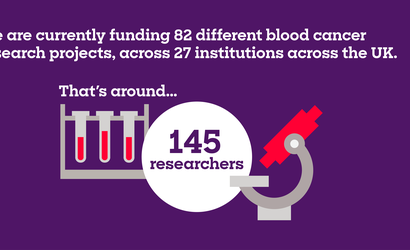
The projects you help us fund
The VIVO Biobank

Funding for researchers

The road to beating blood cancer

Our policy on open access

Our Research Strategy Patient Panel

The Blood Cancer UK Vaccine Task Force
